library(ezGenomeTracks)
#> Warning: replacing previous import 'AnnotationDbi::select' by 'dplyr::select'
#> when loading 'ezGenomeTracks'
#> ezGenomeTracks v0.0.13
#> Easy and flexible genomic track visualization
#> Use citation('ezGenomeTracks') to see how to cite this package
#> For documentation and examples, visit: https://github.com/zmu/ezGenomeTracksSashimi plots are an elegant way to visualize mRNA splicing data, but
few existing tools can do it. Years ago I wrote a custom track class for
pyGenomeTracks and have seen several people using it. Here,
ezGenomeTracks provides a unified ez_sashimi
function that makes it even easier to plot Sashimi plots. It takes a
bigwig file and a link file as inputs.
bw0 <- system.file(
"extdata",
"avg_chr2-231091223_231109786_231113600_0.bw",
package = "ezGenomeTracks"
)
bw1 <- system.file(
"extdata",
"avg_chr2-231091223_231109786_231113600_1.bw",
package = "ezGenomeTracks"
)
bw2 <- system.file(
"extdata",
"avg_chr2-231091223_231109786_231113600_2.bw",
package = "ezGenomeTracks"
)
link0 <- data.table::fread(
system.file(
"extdata",
"chr2-231091223_231109786_231113600_0.sashimi",
package = "ezGenomeTracks"
),
header = F,
col.names = c("seqnames", "start", "end", "name", "score", "strand")
)
link1 <- data.table::fread(
system.file(
"extdata",
"chr2-231091223_231109786_231113600_1.sashimi",
package = "ezGenomeTracks"
),
header = F,
col.names = c("seqnames", "start", "end", "name", "score", "strand")
)
link2 <- data.table::fread(
system.file(
"extdata",
"chr2-231091223_231109786_231113600_2.sashimi",
package = "ezGenomeTracks"
),
header = F,
col.names = c("seqnames", "start", "end", "name", "score", "strand")
)
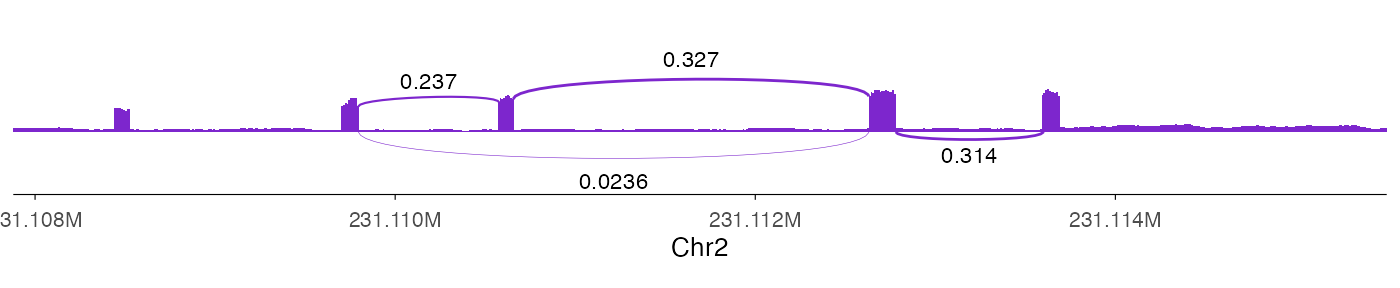
head(link0)
#> seqnames start end name score strand
#> <char> <int> <int> <char> <num> <char>
#> 1: chr2 231109795 231110578 link1 0.23729710 +
#> 2: chr2 231109795 231112631 link2 0.02362313 +
#> 3: chr2 231110655 231112631 link3 0.32665704 +
#> 4: chr2 231112780 231113600 link4 0.31368420 +
ez_sashimi(
coverage_data = bw0,
junction_data = link0 |> dplyr::mutate(score = signif(score, 3)),
linewidth_range = c(0.1, 0.5),
height_factor = 0.03,
region = "chr2:231107879-231115507",
junction_curvature = 0.03
)
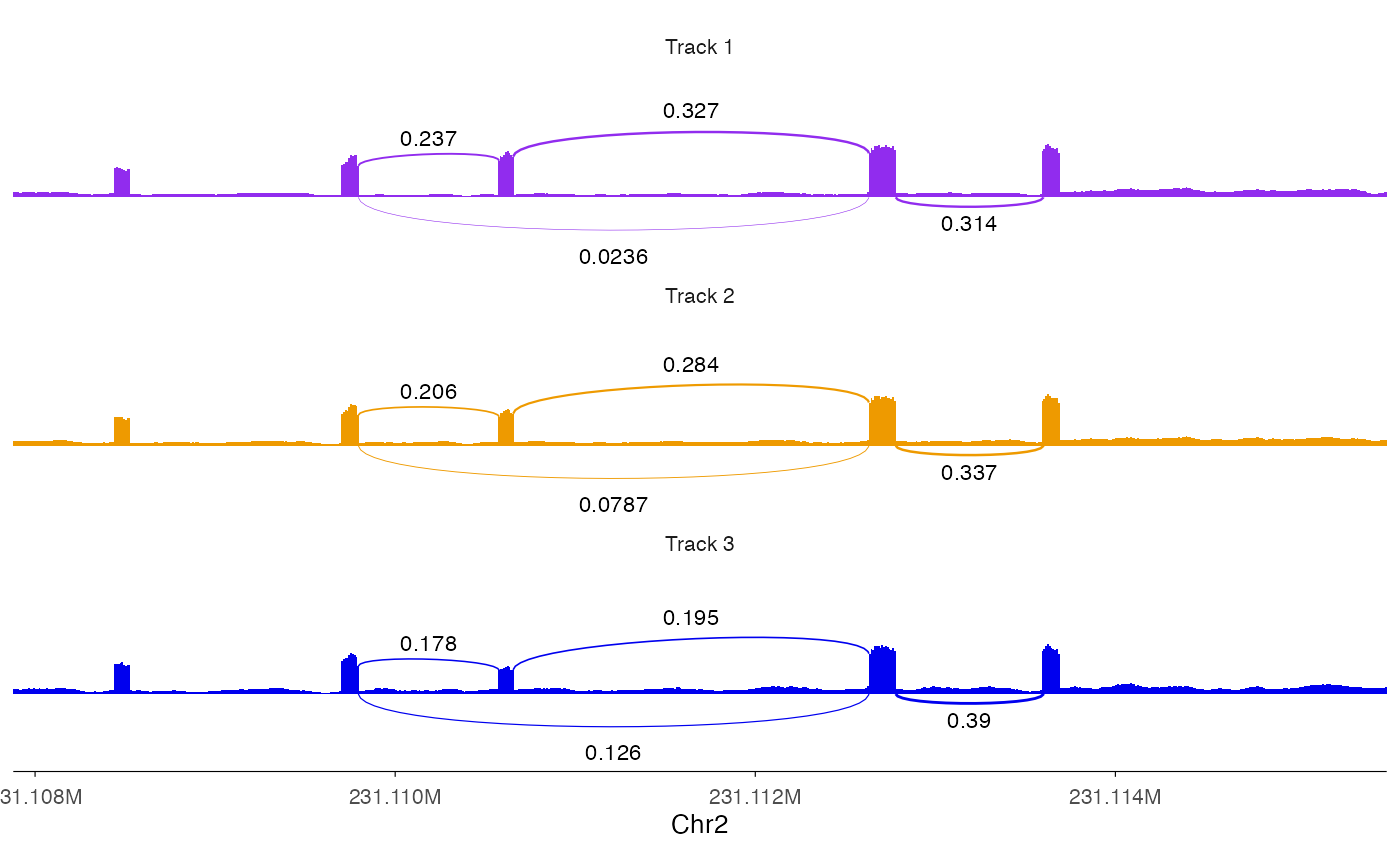
We can plot multiple track together as we do for
ez_coverage.
ez_sashimi(
coverage_data = list(bw0, bw1, bw2),
junction_data = list(
link0 |> dplyr::mutate(score = signif(score, 3)),
link1 |> dplyr::mutate(score = signif(score, 3)),
link2 |> dplyr::mutate(score = signif(score, 3))
),
linewidth_range = c(0.1, 0.5),
height_factor = 0.03,
region = "chr2:231107879-231115507",
junction_curvature = 0.03,
colors = c("purple2", "orange2", "blue2")
)